My research is located in the fields of theoretical chemistry and computational physics. The focus of my work is the development of electronic structure methods for application to large systems and their implementation in program packages optimized for massively parallel computing, such as the codes CP2K and FHI-aims. The focus of my development work are density functional theory (DFT) and many-body perturbation theory (MBPT) approaches to study ground and excited state properties of condensed-phase systems.
Theoretical spectroscopy
Computational spectroscopy has developed into a versatile tool to understand the properties of molecules and materials. My method development of theoretical spectroscopy tools is based on quantum mechanical first principles, in particular Green’s function methods, such as the GW approach. My most recent projects in the field of theoretical spectroscopy are:
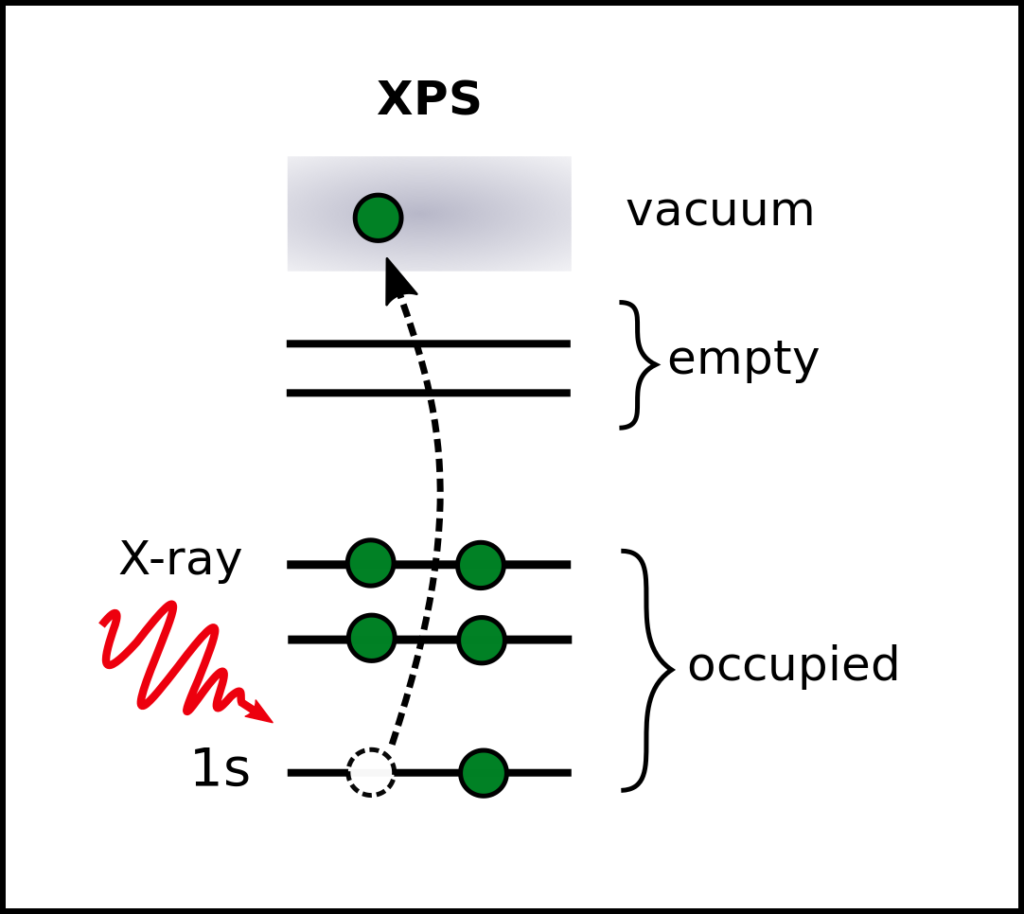
Core-level spectra from GW
Core-level spectroscopy is a powerful tool to characterize molecules, liquids and adsorption processes at surfaces. Accurate computational methods to predict core excitations are important for the interpretation of experimental results. However, the reliable computation of core levels remains a challenge.
Currently, computational core-level spectroscopy is mainly based on Kohn-Sham-DFT. We recently showed that the GW approximation can be used for the accurate prediction of core excitations and can overcome the shortcomings of DFT-based methods.
Related publications:
- D. Golze, J. Wilhelm, M. van Setten, P. Rinke, Core-level binding energies from GW: An efficient full-frequency approach within a localized basis JCTC, 14 (2018), 4856
- D. Golze, L. Keller, P. Rinke, Accurate absolute and relative core-level binding energies from GW JCPL , 11 (2020), 1840
Low-scaling GW for valence states
In canonical implementations, the computational cost of GW scales with O(N4) with respect to system size N, which prohibits application to many systems of interest. I contributed to a low-scaling GW algorithm, which is optimized for massively parallel execution. We applied the low-scaling implementation to technologically interesting applications such as graphene nanoribbons of up to 1734 atoms in size.
Related publications:
- J. Wilhelm, D. Golze, L.Talirz and J. Hutter, C. A. Pignedoli, Toward GW Calculations on Thousands of Atoms, JCPL , 9 (2018), 306
Embedding models for surface systems
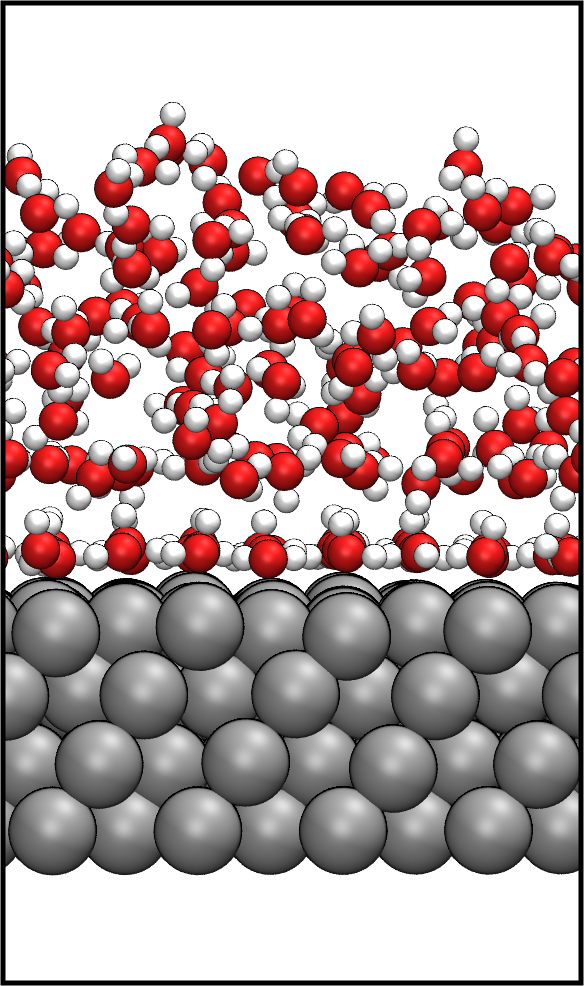
The simulation of complex condensed matter systems, e.g., liquid-solid interfaces requires sampling techniques such as molecular dynamics (MD). However, the timescales accessible in DFT-MD simulations are too small to obtain converged results. Even static calculations remain challenging, in particular when metals are involved. To make such simulations computationally feasible, we developed hybrid quantum mechanics/molecular mechanics (QM/MM) approaches that combine the accuracy of QM methods with computationally inexpensive force fields.
Image-charge augmented QM/MM
My work comprises the development of an image-charge augmented QM/MM model (IC-QM/MM) for the simulation of adsorbates at metallic surfaces. The adsorbates are treated by DFT and the metallic substrate by MM. The interactions between adsorbate and metallic substrate are described at the MM level of theory accounting for induction effects by applying the image charge formulation. We applied this approach to water- metal interfaces, which are relevant for electrochemical applications, and recently in combination with experimental STM images to investigate the growth of organic semiconductors on gold surfaces.
Nano-structured materials
In addition, we developed also non-polarizable QM/MM models for more complex substrates of nano-structured materials, such as the boron-nitride nanomesh generating electrostatic contributions to the force fields from periodic potential fitting.
Related publications:
- D. Golze, M. Iannuzzi, M.-T. Nguyen, D. Passerone and J. Hutter, Simulation of Adsorption Processes at Metallic Interfaces: An Image Charge Augmented QM/MM Approach, JCTC, 9 (2013), 5086
- D. Golze, J. Hutter and M. Iannuzzi, Wetting of water on hexagonal boron nitride@Rh(111): A QM/MM model based on atomic charges derived for nano-structured substrates, PCCP, 17 (2015), 14307-14316
Large-scale DFT calculations
Kohn-Sham (KS) DFT has become the workhorse for electronic-structure calculations. KS-DFT provides a fair compromise between accuracy and computational efficiency enabling the simulation of systems with several hundreds of atoms. However, the computation of large condensed phase systems requires still massive computational resources. To decrease the computational demands of DFT calculations we developed a local resolution-of-the-identity approach (LRI) for mixed Gaussian and plane waves frameworks in combination with computationally efficient integration schemes.
Related Publications:
- D. Golze, M. Iannuzzi and J. Hutter, Local Fitting of the Kohn-Sham Density in a Gaussian and Plane Waves Scheme for Large-Scale Density Functional Theory Simulations, JCTC, 13, 2202
- D. Golze, N. Benedikter, M. Iannuzzi, J. Wilhelm and J. Hutter, Fast evaluation of solid harmonic Gaussian integrals for local resolution-of-the-identity methods and range-separated hybrid functionals, JCP, 146 (2017), 034105